 Nauki ścisłe
Nauki ścisłe
Przełom w badaniach nad śmiertelną chorobą





Chłopcy chorzy na zespół Bartha, genetyczną chorobę o bardzo dużym wskaźniku śmiertelności, w końcu mają szansę na lek. Przyczyniły się do tego badania dr hab. Karoliny Mikulskiej-Rumińskiej, prof. UMK z Instytutu Fizyki. Artykuł o przełomowych wynikach prac międzynarodowej grupy naukowców ukazał się właśnie w "Nature Metabolism".
Zespół Bartha to choroba skorelowana z chromosomem X, dotyka chłopców, dziewczynki natomiast mogą być jedynie bezobjawowymi nosicielkami. Rozwija się w pierwszej dekadzie życia, głównie w pierwszym roku od urodzenia. Występuje z częstością ok. 1 na 400 tys. urodzeń. Poza cechami fizycznymi, takimi jak głęboko osadzone oczy, wydatne policzki, wysokie czoło czy odstające uszy, chorzy cierpią na poważne problemy m.in. z sercem, mięśniami szkieletowymi, nie rosną prawidłowo oraz mają problemy neurologiczne, tj. obniżone zdolności wzrokowo-przestrzenne i ograniczone zdolności matematyczne.
Nadzieja na lek
Chociaż pierwszy przypadek został opisany w 1983 r. przez zespół holenderskiego uczonego Petera Bartha, do tej pory nie udało się znaleźć skutecznego leku. Rokowania pacjentów są bardzo złe, większość chorych umiera we wczesnym dzieciństwie. Jest jednak nadzieja – w artykule "Anomalous peroxidase activity of cytochrome c is the primary pathogenic target in Barth syndrome", który ukazał się w czwartek 23 listopada w "Nature Metabolism", międzynarodowa grupa badaczy i badaczek nie tylko wskazała wiodący mechanizm patogenny, za sprawą którego zespół Bartha się rozwija, ale też wytypowała związek chemiczny, jaki może być wprowadzony do testów klinicznych dla pacjentów z tym schorzeniem. Jedną z głównych autorek publikacji jest dr hab. Karolina Mikulska-Rumińska, prof. UMK z Katedry Biofizyki w Instytucie Fizyki na Wydziale Fizyki, Astronomii i Informatyki Stosowanej UMK.

Awaria w fabryce energii
Do tej pory w terapii chorych skupiano się na leczeniu objawów: problemach z sercem, mózgiem i mięśniami. Nieznany był bowiem mechanizm odpowiedzialny za powstanie choroby, naukowcy nie wiedzieli, co dokładnie wpływa na jej rozwój. Testom klinicznym poddane zostały dwa potencjalne leki, jednak bez sukcesów – jeden z nich nie działa wcale, drugi zaś częściowo łagodzi tylko objawy, ale wciąż nie daje szansy na wyleczenie zespołu Bartha. Niemal 40-osobowa grupa badaczy i badaczek postanowiła przyjrzeć się tematowi kompleksowo. Począwszy od eksperymentów biochemicznych i biofizycznych, poprzez modelowanie komputerowe, a kończąc na testach na zwierzętach oraz badaniach na próbkach z biopsji pacjentów z zespołem Bartha. Prof. Karolina Mikulska-Rumińska była odpowiedzialna za wyjaśnienie mechanizmów molekularnych leżących u podstaw choroby przy użyciu modelowania komputerowego. Wnioski są przełomowe dla sprawy.
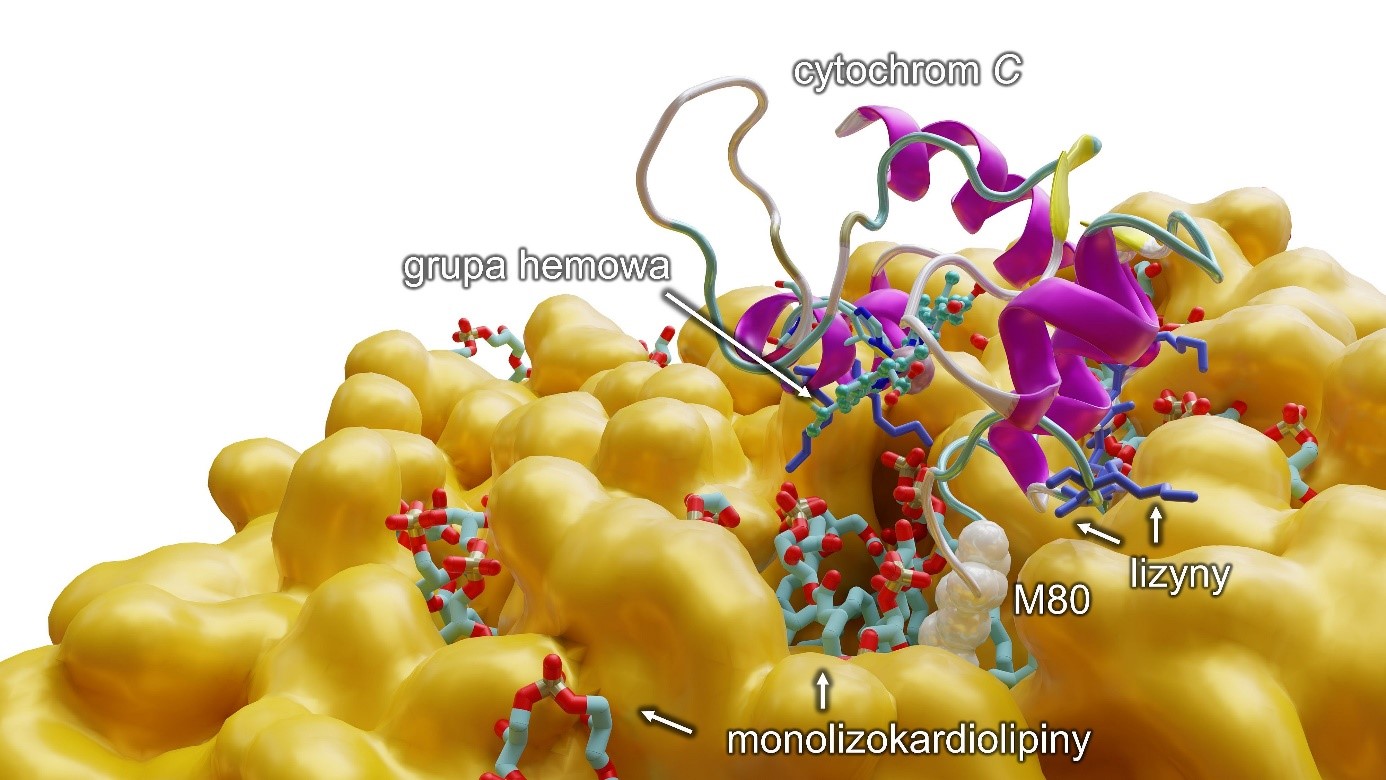
W zespole Bartha mamy do czynienia z mutacją w chromosomie X, a dokładnie w genie TAZ. To gen, który koduje tafazynę, czyli białko, które bierze udział w metabolizmie lipidów, a dokładnie kardiolipin – wyjaśnia prof. Karolina Mikulska-Rumińska. – Co należy podkreślić, kardiolipiny są bardzo ważne - występują w mitochondriach, czyli naszych "fabrykach energii", zasilających wiele procesów w naszym organizmie. Jeśli mamy do czynienia z mutacjami w tafazynie, wówczas kardiolipiny nie powstają, a w zamian akumulują się monolizokardilipiny, czyli kardiolipiny bez jednego łańcucha acylowego. I wtedy mamy poważny problem, ponieważ kardiolipiny wpływają na funkcje stu innych białek mitochondrialnych.
Należy dodać, że kardiolipiny tworzone są z monolizokardiolipin m.in. przy pomocy tafazyny. W zespole Bartha dochodzi do nagromadzenia monolizokardiolipin i deficytu kardiolipin. Co jednak jest bezpośrednią przyczyną dysfunkcji mitochondriów oraz skutków chorobowych określanych jako zespół Bartha, dotąd naukowcom nie udało się wyjaśnić.
Toksyczna maszyneria
Udało nam się ustalić, że monolizokardiolipiny, czyli ta uboższa wersja kardiolipin, łączą się z cytochromem c. To bardzo ważne białko, odpowiedzialne m.in. za oddychanie komórkowe, a także biorące udział w programowanej śmierci komórki – tłumaczy prof. Mikulska-Rumińska. – Odkryliśmy, że monolizokardiolipina, łącząc się z cytochromem c, zmienia jego funkcje, transformując go w anormalną peroksydazę zdolną do katalizowania peroksydacji mitochondrialnych fosfolipidów PUFA. Innymi słowy, kompleks zaczyna działać jak maszyneria tworząca "toksyczne fosfolipidy", które zaczynają wpływać negatywnie właśnie na mięśnie szkieletowe, serce czy mózg. Udowodniliśmy więc, że to ten kompleks - monolizokardiolipiny z cytochromem c jest podstawą, początkiem zespołu Bartha.
Zespół wskazał również konkretny związek chemiczny, który blokuje kompleks cytochromu c z monolizokardiolipiną. Prof. Mikulska-Rumińska odpowiedzialna była za modelowanie komputerowe – pokazała, w jaki sposób ten proces przebiega. Pozwoliło to lepiej zrozumieć podstawy molekularne badanego układu, co jest niezbędne do zaprojektowania efektywnego leku.
Dr hab. Karolina Mikulska-Rumińska, prof. UMK ukończyła studia licencjackie i magisterskie na kierunku fizyka medyczna na Wydziale Fizyki, Astronomii i Informatyki Stosowanej Uniwersytetu Mikołaja Kopernika w Toruniu. Obroniła z wyróżnieniem doktorat z biofizyki oraz habilitację z nauk fizycznych.
Odbyła trzy dłuższe staże naukowe: w firmie farmaceutycznej ADAMED, jako beneficjentka europejskiego stypendium Sciex-NMSch, w Laboratory of Physics of Living Matter na politechnice École Polytechnique Fédérale de Lausanne (EPFL) w Szwajcarii (pół roku) oraz w School of Medicine na Uniwersytecie w Pittsburghu (2,5 roku) pod opieką prof. Ivet Bahar, gdzie m.in. uczestniczyła w projektach National Institute of Health (NIH). Uzyskała m.in. granty PRELUDIUM3, SONATA15 i SONATA BIS12 finansowane przez Narodowe Centrum Nauki (NCN). Jest stypendystką m.in. stypendium habilitacyjnego L'Oréal-UNESCO Dla Kobiet i Nauki (2021), International Rising Talent z międzynarodowego programu For Woman in Science fundacji L'Oréal i UNESCO (2022), nagrody naukowej im. Stefana Pieńkowskiego Wydziału III Nauk Ścisłych i Nauk o Ziemi Polskiej Akademii Nauk (PAN) w dziedzinie fizyki (2022).
Ostanie lata poświeciła na intensywne badania procesu regulowanej śmierci komórki, za które otrzymała Wyróżnienie Rzeczpospolitej Cyfrowej, znalazła się w setce Forbes Women (2021) oraz została nominowana w krajowym plebiscycie wp.pl w kategorii #nauka (2023).





